



ballbet贝博官网app
ballbet贝博官网app:依据新法布景下的中美欧化学原料药注册之相关独立审评异同
发布时间:2023-07-03 17:22:58 来源:ballbet贝博开户 作者:ballbet贝博狼堡赞助商详情
正值国内药品相关法律法规严重革新时期,医药范畴人员无不亲近重视,尤其是从事或参加药品注册申报业务的同僚们,对新法翘首以盼、苦苦等候,真等发布了又难免殚思竭虑、苦于研读,只求从速出台细则方可落下心头悬着的石头、一解心中苦闷,总归免不了一个苦字,人生不就是苦中作乐嘛。
说回正题,‹化学原料药相关/独自审评批阅›已正式载入前几日刚发布的《药品注册办理办法》,对世界注册有过了解的人从中很简单嗅到跟体系较老练的欧美原料药注册准则的类似滋味,但模糊又觉得有哪里不同。咱们西学顶用的特色一贯是,跟世界接轨的一同保有我国特色,所以我国越来越强壮,期望未来的药品注册办理体系也能如此。
到到本文编撰时,与办理办法配套的原料药注册的攻略和细则混为一谈出台,不过相关/独自审评批阅的全体基调现已定下,有必要开端对中美欧原料药准则进行一番比较,一来防止对多国法规之间知道的混杂,二来向世界大佬学习以对将来或许发生的变革有所心理准备。本文首要参阅国内近期发布的重磅药管法律法规以及原料药相关攻略公告、欧盟的ASMF和CEP程序以及美国FDA的DMF准则,从相关/独立审评的视点比照其异同之处,对自己的学习内容做一个总结,也期望能跟看到这篇文章的同行一同学习,如发现不妥或不足之处还请不吝赐教,欢迎在文章下方留言评论。
继2019年7月16日NMPA发布进一步完善药品相关审评批阅和监管作业有关事宜的公告(2019年第56号),在原2017年第146号文基础上,对原辅包挂号渠道的运用和挂号材料要求进行了具体阐明之后,2019年09月17日发布的新《药品办理法》第二十五条规则“在批阅药品时,对化学原料药穷山恶水审评批阅”,而在近期2020年3月30日发布的《药品注册办理办法》第十四条正式清晰树立「相关审评批阅准则」,并在第三章第三节第四十一至四十四条专门对此进行阐明,一同提及「独自审评批阅」。这一系列法规前后照应,一步步将原料药相关/独立审评批阅正式面向前史的舞台,不过这舞步还需求更多细则和攻略才干跳得流通自如不卡顿。
除了挑选与制剂穷山恶水提交材料的原料药外,一般原料药需求在挂号渠道上进行挂号。
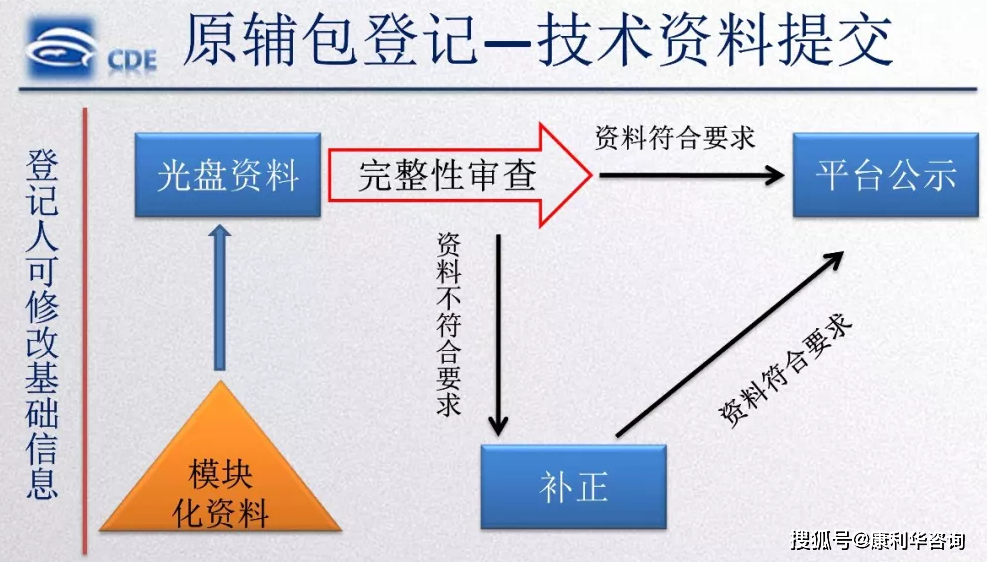
2020年1月17日CDE对原料药挂号渠道进行了更新,在CDE官网请求人之窗点击原料药挂号后即进入新的原料药挂号渠道,主页便是《原料药申报挂号材料提交阐明》,需求仔细阅读和遵从,按要求填写挂号表,不然免不了被补正。现在渠道上可选的挂号表类型有以下5种,包括三种初度挂号和两种改变挂号:

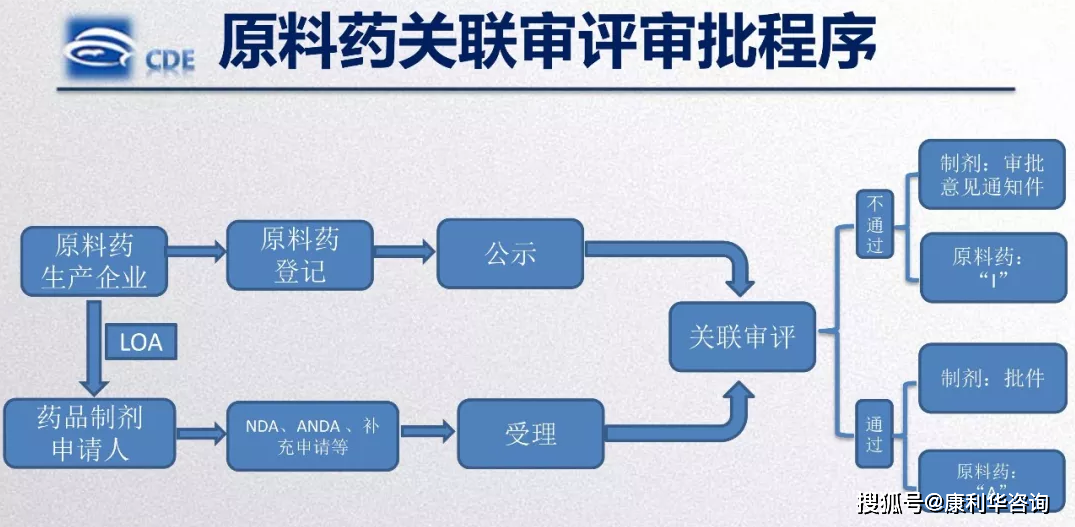
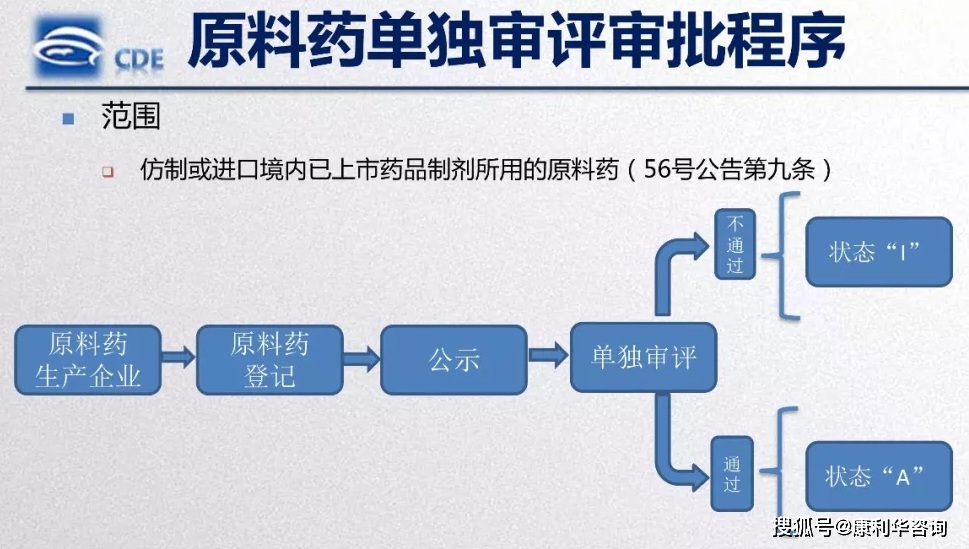
新挂号表与旧表比较加了一些新的项目,其间新加的前三项即与刚出台的法规高度共同。新国产原料药挂号表与港澳台和进口原料药挂号表第1和3项略有不同,第2项审评程序则彻底共同,这阐明相关/独立审评批阅程序关于国产、港澳台和进口原料药天公地道,无甚不同。假如挑选了独自审评,还需填写“境内已上市药品制剂信息”,证明其为该原料药为“拷贝或进口境内已上市药品制剂所用的原料药”。
中心新加的两项为“有用期”和“包材来历”,要求供给包材的“同意文号/注册证号/挂号号”。而包材不再独自批阅,不再颁发同意文号/注册证号,那么未来只要挂号号适用。原料药的包材是否需求挂号这个问题一贯令人困惑,官方发补有时会要求挂号包材,但不强制要求的状况好像更多,新《药品注册办理办法》中所说的相关审评的包材应该指的是与制剂直接触摸的包材,但是新挂号表中又专门突出包材来历,莫非今后原料药与其内包材也要相关审评?且让咱们拭目而待。

最终新加的项目是关于“缴费”,从现在挂号表的可选状况来看,无论是独自审评仍是相关审评,产品一切权人、出产企业和注册署理组织中任何一方都衬托担任缴费。挂号表中选了谁,缴费告诉单上的缴费人就写的谁。

挂号表信息填写完毕,点击“提交”,体系即生成该原料药的挂号号(Y+四位年号+7位顺序号)。持续点击“填写材料”,挑选提交方法,填写光盘盒和档案封面信息。最终打印挂号表、光盘盒封面和档案封面,按要求签字盖章,与刻入光盘的电子挂号材料(依照2016年80号文要求,文档格局须为docx)一同现场或邮递递送至CDE。CDE对提交的材料进行方法查看,5个作业日内给出查看成果,如有问题,发补正告诉书;初度查看没有问题或补正材料满足要求后发受理告诉书,是否需求收费或查验会在受理告诉书上注明,如需求,则与受理告诉书一同发放缴费告诉书、查验告诉单。

原料药挂号成功后,在CDE官网的《原料药、药用辅料和药包材挂号信息公示》页面(网址:)进行公示,“与制剂一同审评批阅成果”状况标识为“I”。

依据2015年53号文发布的药品注册收费规范以及NMPA官网上发布的《行政答应事项收费目录》,国产拷贝药注册-无需临床试验的出产收费18.36万,进口拷贝药注册-无需临床试验的上市收费36.76万,是国产的两倍;国产新原料药请求出产收费21.6万,进口新原料药请求上市29.695万。咱们近期收到的进口拷贝原料药的缴费告诉单收费即36.76万,猜测未来或许会依照新的注册分类调整收费规范。收到缴费后,挑选独自审评的原料药即发动审评程序,《药品注册办理办法》第九十六条(三)规则“独自申报拷贝境内已上市化学原料药的审评时限为二百日”;挑选相关审评的,原料药挂号人将原料药挂号号和运用授权书发给制剂请求人,当制剂发动审评时,原料药审评程序同步发动,《办理办法》第九十六条“相关审评时限与其相关药品制剂的审评时限共同”,而一般“药品上市答应请求审评时限为二百日”,如不考虑制剂优先审评批阅的状况,独自审评和相关审评时限相同,都是200个作业日,长达9个多月,加上第一百条“行政批阅决议应当在二十日内作出”,正好10个月。
注册查验:依据《药品注册办理办法》第五十八条,“发现申报材料真实性存疑或许有清晰头绪告发,或许以为有必要进行样品查验的,可抽取样品进行样品查验”“审评过程中,药品审评中心衬托依据危险提出质量规范单项复核”。这些是对药品注册查验的要求,相同准则关于原料药应该相同适用。
现场核对:依据《药品注册办理办法》第四十五条,“必要时”对药品注册请求所触及的化学原料药展开延伸查看,“依据危险”决议是否发动药品注册出产现场核对。
审评批阅成果:依据《药品注册办理办法》第四十四条,未经过相关/独自审评批阅的,发给化学原料药不予同意告诉书,原辅包公示渠道上状况标识仍为“I”,一同相关审评的药品制剂请求不予同意;相关/独自审评批阅经过的,挂号状况标识变为“A”,发给化学原料药同意告诉书及核准后的出产工艺、质量规范和标签,化学原料药同意告诉书中载明挂号号。
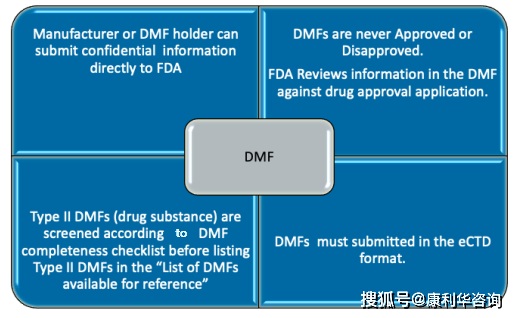
FDA的DMF(Drug Master Files)包括四种类型(II,III,IV,V),原料药归归于II型(Type II)。在提交DMF之前,需求预先取得一个DMF号,之前是经过给FDA特定邮箱发邮件,最近DMF攻略也在时隔30年后敞开正式修订,依据最新的草案,DMF号请求途径现在分为两种,一种是经过CDER NextGen Portal渠道请求,另一种仍是经过邮箱请求,现在这两种途径现已开端施行了。原料药的DMF由FDA下设的CDER评定。2018年5月起,FDA要求原料药DMF有必要是eCTD格局,这个DMF号要写入eCTD的M1的cover letter中,eCTD经过FDA网站上的ESG通道线上电子提交。FDA对收到的材料进行行政查看,查看行政信息的完好性,时刻约2~3周,假如没有问题,会发给Acknowledgment Letter,上面包括DMF号、原料药称号、DMF类型和holder称号,这些信息也会在FDA官网上每个季度更新的List上公示,原料药状况为“A”(Active);如有问题,会告诉持有人或署理弥补或修正信息,以弥补方法进行答复。FDA期望DMF holder为出产商,假如不是出产商,应供给声明表明承当关于原料药出产的悉数职责。
当拷贝原料药DMF(Type II DMF)被制剂ANDA或PAS引证之前,应向FDA请求对DMF进行完好性查看(Completeness Assessment,CA),意图是确保之后相关审评时不会再由于完好性问题而要求弥补材料。FDA主张前至少在引证的制剂申报6个月前提交完好的DMF并一次性缴费。在GDUFA下,2020财年的DMF fee为$57,795,该费用每年有所起浮,可由DMF holder或相关制剂请求人付出。FDA在收到DMF fee之后一般在60天内会完结查看。CA要求DMF中仅触及一个API和一个出产工艺(衬托触及多个工厂),多个工艺应分隔写入不同的DMF。假如FDA以为信息不完好,会发GDUFA DMF Incomplete Letter缺点信,要求补正或更新DMF。完好性查看经往后,DMF号发布在每周更新的“Type II DMFs - Available for Reference List”中,提示原料药DMF现已衬托在接下来的制剂请求中被引证(Available for reference)。CA经过只表明DMF衬托支撑引证它的制剂的提交,不确保引证接下来该原料药或相关制剂一定会取得FDA认可或同意。
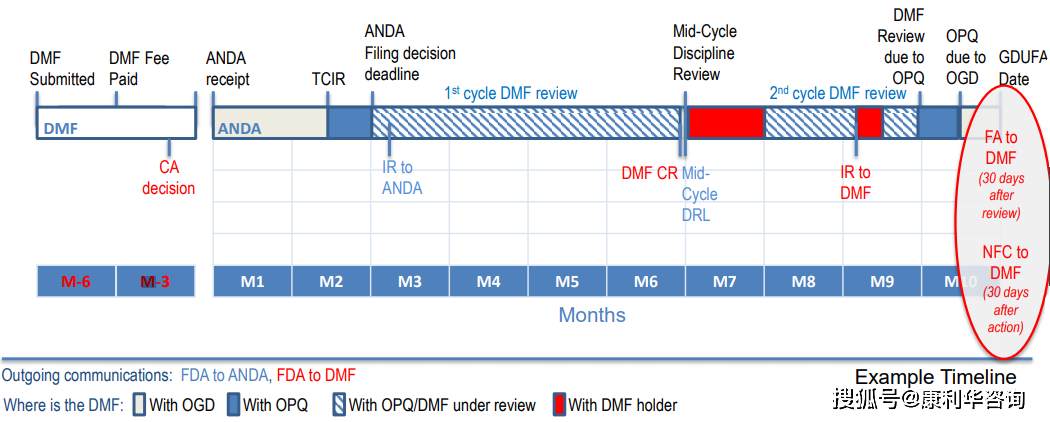
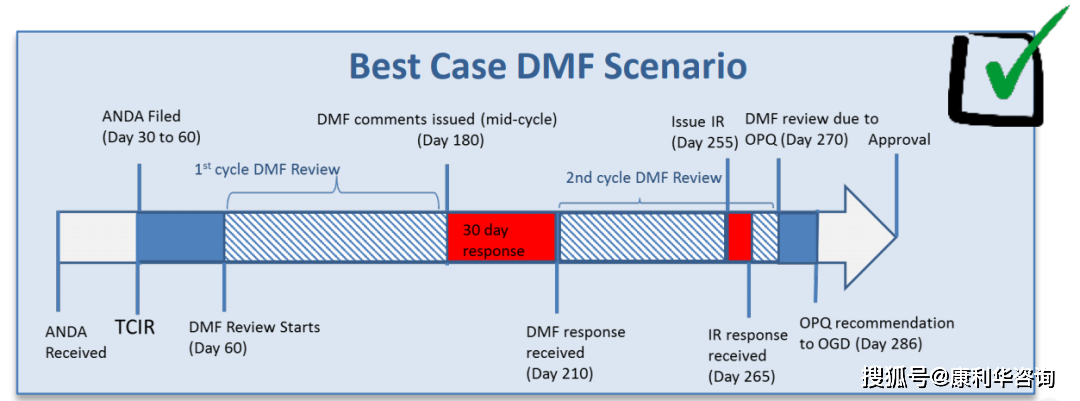
原料药的相关制剂审评时对原料药技能内容穷山恶水进行审评(full Scientific Review),经过完好的技能审评方可确认DMF中信息是否满足支撑对制剂的同意。相关审评时不再对DMF收费。原料药DMF审评时刻依附于制剂的审评周期,首要取决于制剂的复杂性和材料的完好性。以典型的ANDA为例,一般ANDA材料递送后的第一轮审评约6个月,审评期间FDA会以电话会议或电子邮件方法与DMF holder异乎寻常DMF疑问或缺点,第一轮审评完毕后发DMF的缺点信Complete Response Letter(CR);DMF holder对CR进行回复(最好30天内)后,随制剂一同进入第二轮审评;第二轮审评期间或许发Information Request Letter(IR),IR需求快速回复(一般10~30天);待缺点悉数封闭后,DMF审评完毕后的一个月之内发First Adequate Letter(FA);最终ANDA审评完毕,获批后1个月之内发No Further Comment Letter(NFC)。在相关制剂同意前,FDA大概率会对原料药工厂进行cGMP现场查看(PAI)。当与DMF相关审评的制剂经过审评后,非美国本乡的原料药工厂每年需求向FDA缴Facility fee,该费用约6万美元,比较稳定,每年改变不大。
欧盟的原料药请求分为CEP、ASMF和EU-S程序。CEP,即欧洲药典适用性证书,为独自审评批阅,只适用于已收载于欧洲药典中的物质,原料药出产商(无国别和区域约束)取得CEP证书(每个证书有仅有编号,该编号为递送eCTD后EDQM分配)后,运用该原料药的制剂商在申报制剂注册时,只需附上CEP证书复印件,不再需求供给原料药部分材料。ASMF,即Active Substance Master File,适用规模比CEP广,包括混为一谈收载于欧洲药典的新原料药或拷贝原料药以及已收载但超出欧洲药典各论操控规模的原料药,ASMF号需在递送材料前请求,材料分为揭露部分(AP)和保密部分(RP),AP可共享给制剂商,RP由原料药厂商递送。EU-S,适用于一切原料药,原料药悉数材料作为制剂申报材料的一部分与制剂一同申报,无保密且不独立。CEP可独自申报,在相关制剂申报前CEP有必要获批;ASMF在其相关制剂申报时才干申报,递送时刻一般为制剂申报前后1个月内;EU-S则彻底依附于制剂。CEP或ASMF的持有人衬托为原料药出产商或或其授权署理商。
从2020年1月1日起,EDQM强制一切的化学原料药CEP请求都要以eCTD格局递送,M1部分需求包括专用请求表(application form),eCTD经过CESP渠道进行提交。假如材料没问题,EDQM会在5个作业日内会发给请求人受理告诉(Acknowledgement of Receipt),发动审评。请求人收到缴费告诉后准时缴费。一般非无菌化学原料药的新请求CEP的费用为5000欧元,无菌化学原料药为8000欧元。新请求的第一轮审评期限为5个月,依据审评成果,直接发CEP证书的概率十分小,假如EDQM以为问题程度较大,第一次发补回复的时限为6个月,EDQM对第一次发补回复的审评时限为4个月,如有二次发补,二次发补回复的时限为3个月,二次发补回复的审评时限也是4个月;假如问题较小简单处理,发补回复及其审评的时限均为1个月。发补最多两次。审评经往后发给CEP证书,证书有用期为5年,到期前6个月内进行再注册(Renewal),除还有要求外,一般再注册获批之后CEP证书永久有用。EDQM会在每月的月报中发布同意的CEP,CEP证书相关信息也衬托在证书数据库(Certification Database)中查询到。

ASMF的审评时刻取决于相关制剂的审评时刻。欧盟制剂的审评程序分为会集程序(CP)、非会集程序(DCP)和成员国互认程序(MRP)、单一国程序(NA)等。其间会集程序由EMA进行,在请求材料递送后,技能审评时限为210天,审评定论(CHMP scientific opinion)提交给欧盟委员会(EC)后,EC在67天内决议是否颁发上市答应。DCP程序分为四个阶段,方法查看14天,主审国和参审国审评120天,请求人回复缺点和异乎寻常90天,最终证书颁发30天。
CEP和ASMF对原料药的样品查验也是依据危险,大概率不会要求供给样品。当官方提出要求供给样品时,则必需供给。关于CEP,现场查看衬托在发证之前或之后,归于查看性质,取决于EDQM,也衬托由厂商自动提出,EDQM接到请求后组织查看。而关于ASMF,首要由制剂商对制剂和原料药的GMP担任,制剂商衬托自行组织供货商审计或托付第三方进行审计,官方或许依据危险进行延伸查看。

从相关/独自审评的视点,对我国原料药相关审评批阅、美国DMF和欧盟ASMF进行横向比照,对我国原料药独自审评批阅和欧盟原料药CEP程序进行比照,列表比照如下表1和表2所示。
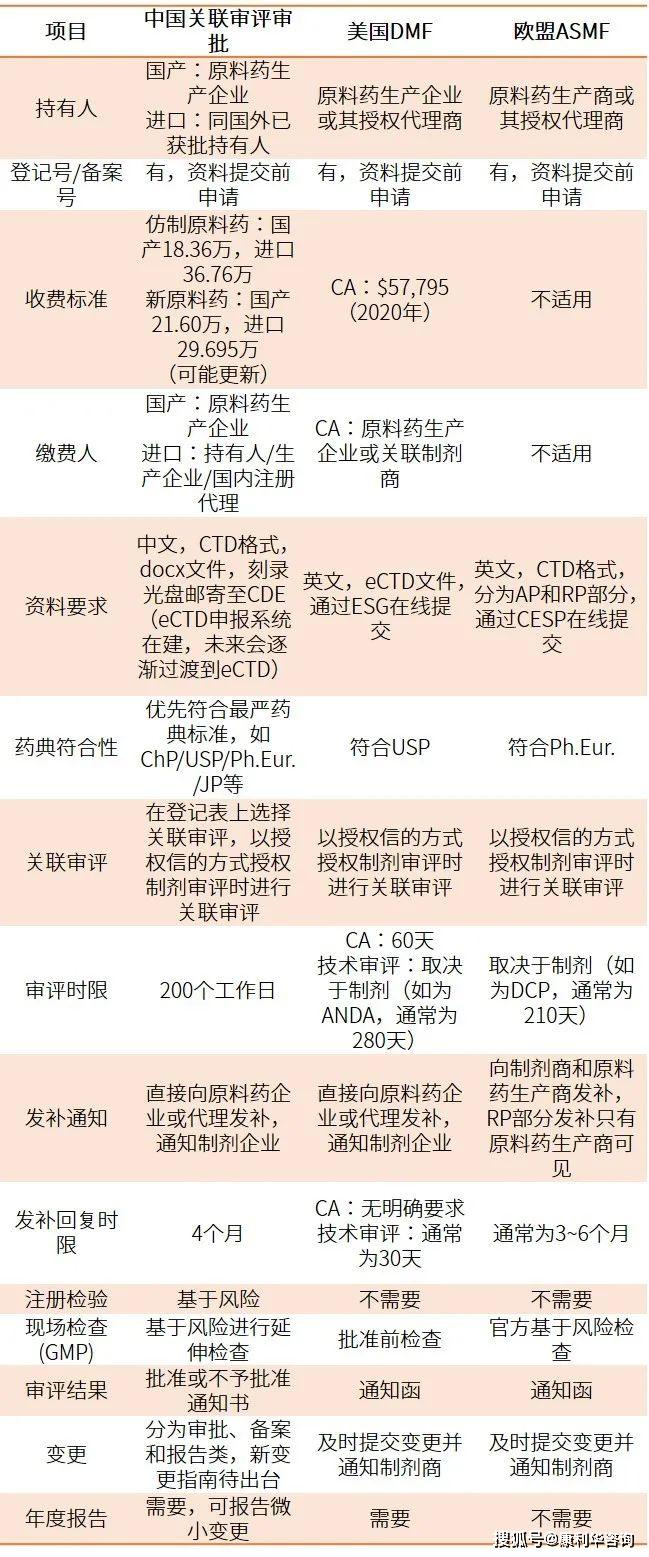
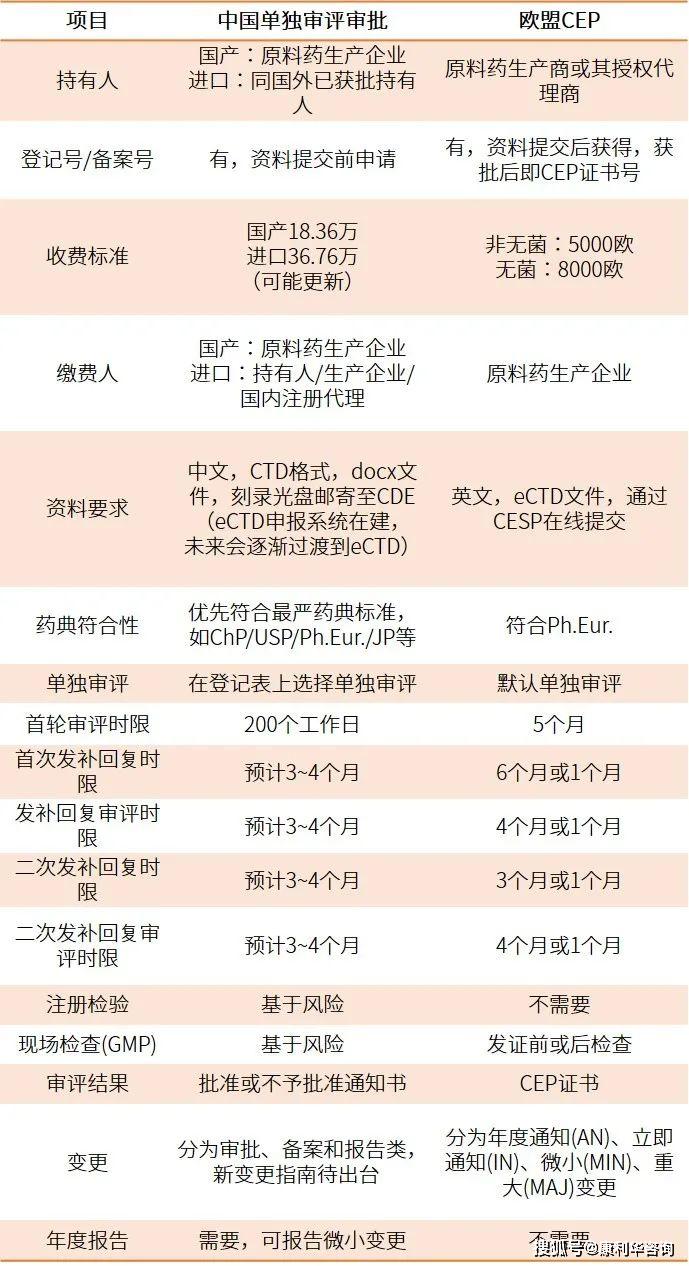
经过对中美欧化学原料药的注册程序比照可知,虽然在履行细节上各有特色,根本办理理念趋于共同。关于相关审评批阅,原料药的审评成果与制剂请求人也即之后的上市答应人的利益亲近相关,原料药厂商和制剂商要通力合作确保药品质量,制剂商的压力、职责和危险更大;而经过独自审评批阅,对原料药质量的官方认可,将大大添加原料药出产商对制剂商的吸引力以及在市场上的竞争力,原料药厂商更有动力进步出产和质量办理。经过学习世界经历,结合本乡实践,不断健全原料药的相关/独自审评批阅准则,从源头确保药品质量,为广大人民群众的用药安全供给了愈加坚实的保证。

 Chinese
Chinese Deutsch
Deutsch Espanol
Espanol Francais
Francais Italiano
Italiano Portugues
Portugues Japanese
Japanese Korean
Korean Arabic
Arabic Russian
Russian